
JDFTx恒电势计算方法与应用
JDFTx恒电势计算方法与应用
简要介绍:
JDFTx是一种平面波密度泛函理论(DFT)代码,旨在易于开发和使用。
JDFTx是使用高度模板化和面向对象的C++11代码编写的,目的是在DFT++代数框架中表达所有物理特性,同时保持较小的内存占用并支持一系列硬件架构(如使用CUDA的GPU),而不需要为每种架构手动优化实现。
相比于最流行的DFT软件VASP所需要的输入文件:
1 | INCAR # 输入文件 设置计算参数 |
JDFTx 所有命令的设置都可以只写在一个输入文件command.in,JDFTx 支持把结构文件直接以命令的形式写在输入文件里
输入文件是一个命令列表,每行一个;
命令可以按任何顺序出现;根据你的喜好将它们分组;
#后一行中的所有内容都被视为评论并被忽略;
空格分隔单词;多余的空格将被忽略;
\ 号表示续行。
其中最基本的结构输入格式如下:
1 | command <value> |
以Si为例的布里渊区采样
硅具有金刚石结构,立方晶格常数为5.43埃(10.263玻尔半径)。每个立方晶胞包含8个硅原子(位于顶点、面心和两个半晶胞体心)。但为了更简洁地描述周期性,我们可以使用更小的面心立方晶胞,仅包含2个硅原子:
1 | # ----------------POSCAR---------------- |
将以上内容保存为Si.in,运行:
1 | jdftx -i Si.in | tee Si.out |
电子密度可视化,运行以下命令生成XSF文件:
1 | createXSF Si.out Si.xsf n |
可以用VESTA打开Si.xsf:
进阶
布里渊区采样对能量的影响
修改Si.in中的k点折叠命令:
1 | kpoint-folding ${nk} ${nk} ${nk} |
编写批量计算脚本run.sh:
1 |
|
运行后得到能量收敛结果:
1 | -7.317807660097655 Si-1.out |
当k点密度达到8×8×8时,能量已收敛至0.0001 Hartree以内。
Monkhorst-Pack网格对比
在Si.in中添加:
1 | kpoint 0.5 0.5 0.5 1. |
重新运行脚本,得到结果:
1 | -7.849461799239376 Si-1.out |
Monkhorst-Pack网格收敛更快,但对称性简化的k点数(nStates)更多,计算时间更长。
溶液中的分子计算:溶剂化模型入门
化学反应常发生于溶液中,而利用DFT预测反应机理需计算溶液中分子的结构和自由能。直接通过溶剂分子进行全原子描述需数千次DFT计算以采样所有溶剂构型,计算量极大。实际可行的替代方案是使用连续介质溶剂化模型,直接近似溶剂对溶质的平衡效应,而无需处理单个溶剂分子的构型。
JDFTx的核心开发方向之一,是在联合密度泛函理论(Joint Density Functional Theory,JDFT)框架下构建从连续介质到经典DFT的多层次溶剂化模型(JDFTx的“J”即来源于此)。本教程演示如何将分子置于溶液中,并介绍控制溶剂化和流体性质的相关命令。
这里介绍一下 include 命令,这个命令就相当于 latex 的 \input 命令,include 命令可以把一个文件的内容包含到当前文件中。
借用此命令,我们可以把一些公共的设置放在一个文件中,然后在其他文件中引用它们。这样可以避免重复编写相同的代码。也可以把冗长的结构文件单独放在一个文件中,然后在主输入文件中引用它们。
以水分子溶于液态水为例。该过程的自由能与液态水的蒸气压相关,实验测量值为 -0.0101 Hartrees(约 -6.33 kcal/mol)。
首先进行真空中的计算:
公共输入文件 common.in:
1 | # 保存为 common.in |
真空计算输入文件 Vacuum.in:
1 | include common.in # 相当于把 common.in 复制到这里 |
保存为 Vacuum.in,运行:jdftx -i Vacuum.in | tee Vacuum.out
此计算将输出真空中水分子的优化几何结构和波函数。
溶液计算(LinearPCM 模型)
基于真空计算结果进行溶液化计算:
LinearPCM输入文件 LinearPCM.in:
1 | include common.in |
保存为 LinearPCM.in,运行:jdftx -i LinearPCM.in | tee LinearPCM.out
1 | # Energy components: |
输出文件中:
Linear fluid 段落表示流体自由度优化(通过线性Poisson-Boltzmann方程求解)。
能量项新增 Adiel,表示溶剂的贡献。
溶剂化自由能 = LinearPCM.out 最终能量 - Vacuum.out 最终能量。
本例结果为 -0.0113 Hartrees,与实验值 -0.0101 Hartrees 吻合。
通过以下命令可视化溶剂中的束缚电荷密度:
1 | createXSF LinearPCM.out LinearPCM.xsf nbound |
相比于VASPSol,JDFTx 提供了更多的溶剂化模型包括:
- LinearPCM
- NonlinearPCM
- SaLSA
- ClassicalDFT
其中LinearPCM或NonlinearPCM又有很多variant,以确定空腔和相关能量(cavitation, dispersion等)。
变体包括:
- CANDLE
- SCCS
- GLSSA13 (VASPsol)
- …
常用溶剂化模型对比
| 模型 | 特点 | 命令示例 |
|---|---|---|
| LinearPCM (GLSSA13) | 线性响应,腔体由溶质电子密度定义,广泛用于固态计算(如VASPsol) | fluid LinearPCMpcm-variant GLSSA13 |
| SCCS | 自洽连续介质溶剂化模型(Quantum Espresso实现),腔体定义方法与参数不同 | fluid LinearPCMpcm-variant SCCS_* |
| CANDLE | 最新推荐模型,结合非局域腔体定义与局部介电响应,修正电荷不对称性 | fluid LinearPCMpcm-variant CANDLE |
| NonlinearPCM | 非线性介电/离子响应,适用于电化学电容计算 | fluid NonlinearPCMpcm-variant GLSSA13 |
| SaLSA | 非局域角动量展开响应,参数少但计算耗时(约10倍于线性模型) | fluid SaLSA |
| ClassicalDFT | 全原子经典DFT描述溶剂微观结构,计算耗时极高(约100倍于线性模型) | fluid ClassicalDFT |
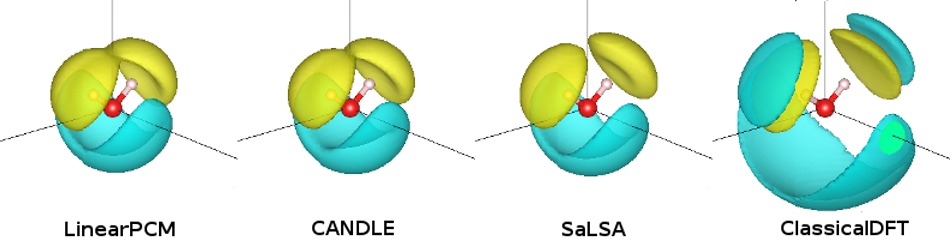
电荷可视化:通过 createXSF 生成各模型的 nbound 电荷密度,观察:
LinearPCM、CANDLE、SaLSA 的电荷集中在溶质表面。
ClassicalDFT 显示多层壳状溶剂结构。
带电表面与电化学系统计算
结合电解质(溶剂+离子)的连续介质溶剂化模型,我们可以直接处理溶液中的带电表面,这对电化学系统尤为重要。电极表面通过外部电路设定电子化学势(μ),电子数量随μ平衡,导致表面带电(除零电荷电位PZC外)。
本教程以Pt(111)表面的电荷-电势曲线计算为例,演示恒电势计算(通过target-mu命令直接设定电子化学势)。
以下是关于 target-mu 命令的详细解释:
1 | target-mu <mu> [<outerLoop>=no] |
<mu>:设定电子化学势的绝对值(以Hartree为单位,相对于真空能级)。
此命令用于恒电势计算(固定化学势μ,而非固定电荷数),模拟电化学系统中电极与外部电路接触时的电子交换行为。
化学势 <mu> 的设定
μ是绝对量,需根据参考电极校准。例如:
相对于标准氢电极(SHE)的电势V(单位:伏特)转换为:
1
mu = -(Vref + V)/27.2114
`Vref`:SHE相对于真空能级的电位(实验值约4.44 V)。
`27.2114`:Hartree与eV的转换系数(1 Hartree ≈ 27.2114 eV)。
示例:若需模拟相对于SHE为+0.5 V的电位:
1
target-mu -(4.44 + 0.5)/27.2114
物理背景
巨自由能(G):
恒电势下系统的热力学势为 G = F - μN,其中 F 是亥姆霍兹自由能,N 是电子数。此量在固定μ时具有变分最小性。
与固定电荷计算的差异:固定电荷时优化 F,而固定μ时优化 G,更贴近真实电化学界面条件。
设置target-mu后将会直接进行巨正则系综变分优化:μ全程固定,电子数连续调整。
输出文件中的自由能标记为 G(巨自由能),而非固定电荷时的 F。
电子数 N 会在迭代中动态更新,直至与μ平衡。
公共输入文件 common.in:
1 | # 保存为 common.in |
中性表面计算
输入文件 Neutral.in:
1 | include common.in |
运行:
1 | jdftx -i Neutral.in | tee Neutral.out |
中性计算结果为后续带电表面计算提供初始状态和PZC(零电荷电位)。
恒电势计算
输入文件 Charged.in:
1 | include common.in |
使用Bash脚本批量运行不同μ值计算:
1 |
|
保存为run.sh,运行:
1 | chmod +x run.sh |
脚本将μ从-1 eV到+1 eV(相对于PZC)分21步计算,每次调整0.1 eV。首次计算耗时较长,后续计算因继承前次状态加速。
1 | # Energy components: |
这里是计算后的一个输出文件,其中G就是自由能。
数据分析与可视化
- 提取电荷-电势数据:
1 |
|
保存为collectResults.sh,运行:
1 | chmod +x collectResults.sh |
- 使用gnuplot绘制电荷密度曲线:
1 | A = 2*((5.23966*0.529e-8)**2)*sin(pi/3) # 表面积(cm²,上下表面) |
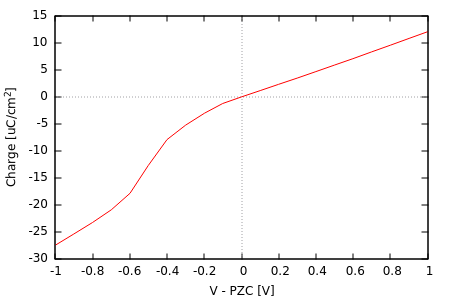
结果将显示Pt(111)表面电荷随电势变化曲线,正负区斜率差异反映CANDLE模型的电荷不对称性。
本文主要讲述了 JDFTx 基本的用法、溶剂化和固定电势模拟,相比于 VASP,JDFTx 有以下优势:
- 提供变量支持,提供更灵活的操作
- 支持 GPU 加速,加速效率很客观
- 提供更多的隐式溶剂化模型
- 内置了恒电势算法,通过
target-mu命令指定电势后,JDFTx 将会固定$\mu$自动优化电子数到指定值。
缺点:
- 计算速度比VASP慢
最后,本文内容全部来自于官网,官网还提供了非常详细的例子和文档,感兴趣的同学可以去看看。